Data analysis III: Biological NMR
生物学作业代写 Make sure that you provide a detailed figure legend and cite the figure in the main text. Try to make your figure as clear and simple as possible.
Nuclear Magnetic Resonance is a unique and extremely powerful biophysical technique that provides detailed information about individual atoms in biological macromolecules. NMR can be used to solve a variety of biological and biomedical problems, including characterization of:
– Structure and dynamics for macromolecules (proteins and nucleic acids) including different functional states and short-living intermediates in vitro and in vivo (inside the living cells)
– Functional conformational changes and flexibility of macromolecules
– Allosteric pathways and mechanisms of signal transduction through protein structure
– Intrinsically disordered proteins (IDP)
– Molecular mechanisms of protein folding, misfolding and aggregations
– Ligand and drug binding including characterisation of binding sites and molecular contacts as well as thermodynamics and chemical kinetics of binding
– Protein-protein and protein-nucleotide interactions including transient multicomponent interactions
This project is primarily designed to introduce you to this state-of-the-art technique and illustrate some practical aspects of modern protein NMR spectroscopy. Each section provides a brief introduction to the topic and several examples. You will need to answer 2 NMR questions as a part (50%) of Data analysis III: Biomolecular NMR spectroscopy and Mass Spectrometry assessment that forms 25% of the overall module mark.
Where can I learn what is NMR? 生物学作业代写
Prof Breeze – NMR Lecture (Minerva)
BioNMR@Leeds facility tour:
https://mymedia.leeds.ac.uk/Mediasite/Play/3acfe2000a364bd3a49ea7a00fb8217c1d
Other useful links:
Short intro to NMR:
Short intro in bioNMR from leading experts in the field:
Where can I learn more about NMR (not required for this module)?
Lectures by James Keeler: http://www–keeler.ch.cam.ac.uk/lectures/
Lectures from James Keeler are also available on YouTube: https://youtu.be/nM7jQFhrvR0
Part 1: Prediction of protein secondary structure
Background information:
Protein secondary structure is the local spatial arrangement of a polypeptide’s backbone atoms without regard to the conformations of its side chains. Protein secondary structure prediction is often the first step toward tertiary structure prediction. It can also provide information about protein stability, activity, and functions.
Backbone NMR chemical shifts can be used to accurately identify the location of secondary structures in a protein. Positions of individual peaks in the NMR spectrum (i.e., chemical shifts) are extremely sensitive characteristics of individual bonds as they depend on a local microenvironment for a corresponding group of atoms and thus, provide information about site-specific structural and dynamic features of proteins.
In folded proteins, different atoms have very different microenvironments.
As a result, different residues from a folded protein usually have very different chemical shifts. On the contrary, for unfolded proteins variations of microenvironments are much smaller and chemical shifts only depend on the local chemical environment, i.e., amino acid types of the residues itself and its adjacent residues. These chemical shifts can be directly calculated from the protein sequence using suggested random coil (fully unfolded) chemical shifts. In the folded protein, chemical shifts strongly correlate with local protein structure. By comparison experimentally observed chemical shifts to a set of residue-specific random coil (fully unfolded) chemical shifts, the location and the type of protein structure can be identify using chemical shift index (CSI) calculations. 生物学作业代写
This method assigns significant downfield secondary shifts (when the chemical shift is significantly greater than the residue-specific random coil reference value) to ‘+1’s’ , upfield secondary shifts (when the chemical shift is significantly smaller than the residue-specific random coil reference value) to ‘ -1’s’ and small secondary shifts (when the chemical shift is significantly is similar to the residue-specific random coil reference value) to ‘0’s’ . A simple graph showing CSI vs. residue numbers can be then generated. Any group of four or more -1 is assigned to an a–helix; any group of three or more +1 is assigned to a b–strand. All other combinations are assigned as coil:
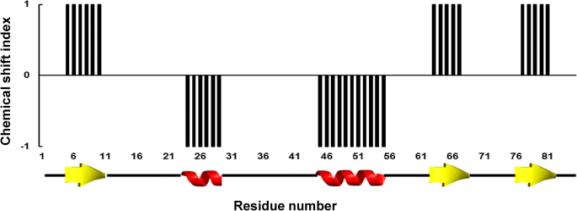
Question 1 (25 marks in total): 生物学作业代写
Using the CSI method, predict secondary structure in the nucleotide-binding domain (NBD) of bacterial Hsp70 (DnaK) using backbone NMR assignments of the protein in the nucleotide-unbound state. Compare the CSI secondary structure prediction with secondary structure predicted from the NBD amino acid sequence using the PSIPRED algorithm.
Show and explain with the use of illustrations the results of this analysis; highlight the most important differences in DnaK secondary structure predictions obtained using these two approaches; briefly discuss key advantages and limitations of CSI and PSIPRED.

Instructions:
1) Download backbone chemical shifts for the protein from Biological Magnetic Resonance Data Bank, BMRB, https://bmrb.io/ (BMRB accession code: 17209). Make sure that you save an NMR-STAR v3 text file on your local computer. This file contains information about the system, experimental conditions and backbone chemical shifts.
2) Use the CSI3.0 server (http://csi3.wishartlab.com/cgi-bin/index.php) to predict secondary structure from backbone NMR chemical shifts of NBD. Upload the NMR- STAR v3 file from step 1 to the server and submit the job.
3) Use the PSRPRED server (http://bioinf.cs.ucl.ac.uk/psipred/) to predict secondary structure from the protein sequence (UniProt accession code P0A6Y8). Follow the instructions on the website.
Note: You should only compare secondary structure for the nucleotide-binding domain of DnaK (residues 1-388).
Presentation of your results: Show a graph(s) with secondary structure elements obtained by CSI and PSPRED methods vs. protein residue numbers; highlight regions with the most significant differences between two algorithms. You do not need to highlight and discuss each individual change and each individual residue. Instead, you need clearly identify whether different methods predict similar or different secondary structure elements for different parts of the protein. You only need to compare the position of loops (unstructured regions), b- strands and a-helices. You can copy/paste and align graphs obtained from CSI and PSPRED online tools or (if you prefer) make you own graph(s). Make sure that you provide a detailed figure legend for each figure and cite all figures in the main text. Try to make your figures as simple as possible while still providing a clear message. 生物学作业代写
The text for this answer (excluding figures) should NOT exceed 1 side of A4.
Part 2: Chemical shift perturbation (CSP) analysis
Background information:
The CSP analysis is an extremely sensitive tool to follow changes in protein structure and dynamics upon any local or global conformational changes in a protein. Any local perturbations (e.g., ligand binding, amino acid substitution, protonation of a side-chain, conformational changes, etc.) within ~5-7 Å from the atom of interest usually result in chemical shift perturbations. As a result, the CPS analysis is a very simple but extremely informative experimental technique to study ligand binding, chemical reactions, pH titrations of functionally important side chains, and characterization of allosteric (long-range) changes in the protein structure.
To perform the CSP analysis, two or more 2D amide spectra (HSQC) are usually recorded, for example:
(i) in the presence and in the absence of a ligand OR
(ii) at different pH
OR
(iii) at different temperatures.
Any changes in peak positions in the NMR spectra can be then monitored with high accuracy. Finally, significant CSPs can be mapped into protein structure (if available) to identify regions affected by a process (e.g., ligand binding, pH changes, temperature). 生物学作业代写
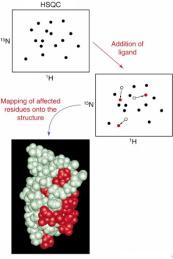
| Illustration from Prof Breeze’s NMR lecture |
Question 2 (25 marks in total): 生物学作业代写
Hsp70 chaperones are key hubs in cellular proteostasis and protein quality control networks in the cytoplasm and organelles. They facilitate protein folding, disaggregation of misfolded proteins, protein degradation, protein refolding and assembly of protein complexes. Hsp70 functions and activity rely on ATP binding to the Hsp70 N-terminal nucleotide-binding domain (NBD) to allosterically alter substrate affinity to the C-terminal substrate-binding domain (SBD). As the Hsp70 family is a putative drug target, initial screening of the NBD of Hsp70 reveals a compound A that inhibits chaperone ATPase activity with Ki of ca. 50 µM.
To obtained molecular details of interactions between NBD and compound A, 2D amide spectra of 100 µM of 2 H,15 N labelled isolated NBD of Hsp70 were recorded in the absence and present of 5 mM of the compound A. Using the chemical shift tables provided in Question2_CSP.xlsx (Minerva),perform the chemical shift perturbation analysis of Hsp70 NBD; based on this analysis, suggest a binding site(s) and plausible mechanism(s) of action for the compound A; suggest an independent experiment(s) to test your suggestions.
Instructions:
1.Using1 Hand 15 N chemical shifts recorded in the presence and absence of compound A (Question2_CSP.xlsx), for each NBD residue calculate total chemical shift perturbations (CPSs) using the following equation:
![]()
where ΔCS(1H) is the difference between proton chemical shifts recorded in the absence and presence of compound A and ΔCS(15N) is the differences between nitrogen chemical shifts recorded in the absence an presence of compound A. 生物学作业代写
2. PlotCSPsas a function of residue numbers and clearly identify residues with significant (>0.2 ppm) CSPs. Attach this plot to your report. Make sure that you provide a detailed figure legend and cite the figure in the main text. Try to make your figure as clear and simple as possible.
3. UsingPyMol, mapresidues with significant CSPs into 3D structure of NBD to identify the binding site of the compound A (PDB accession code: 2KHO.pdb; the nucleotide-binding domain, NBD, corresponds to residues 1-388 in this structure). To identify the binding site and suggest a plausible mechanism(s) of action, try to answer the following questions: Why only some peaks are affected by binding to the compound A? Are all residues with significant CSPs located close together? Are residues with significant CSPs located near functional sites of the protein?
The text for this answer (excluding figures) should NOT exceed 1 side of A4.
更多代写:新西兰管理代写 sat考试作弊 新加坡留学生作业代写 艺术学essay代写 音乐学论文代写 微处理器系统代写
合作平台:essay代写 论文代写 写手招聘 英国留学生代写



